
Medical Devices Regulation (MDR)
What contains MDR?
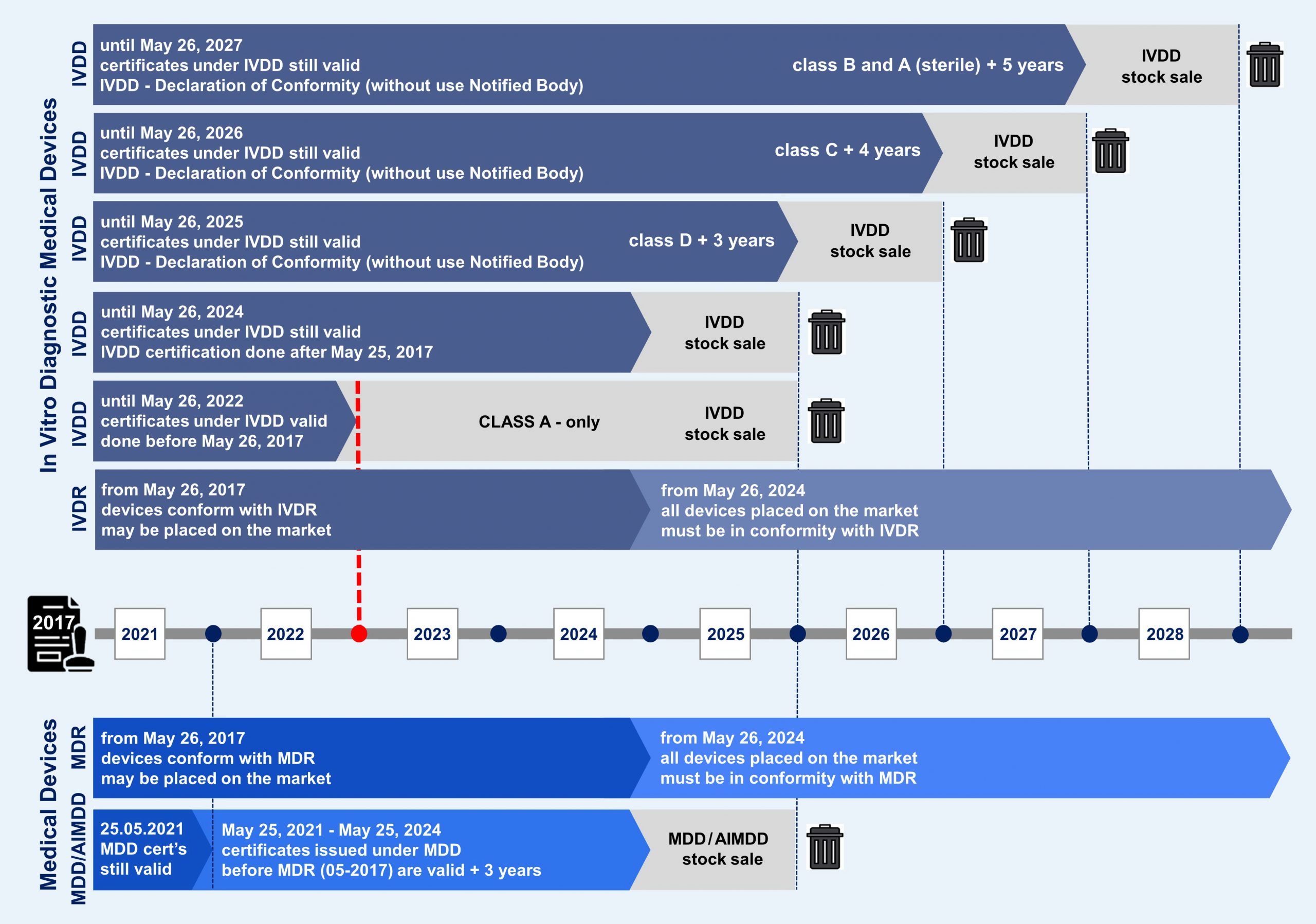
Transition Timelines
The new Regulations create a robust, trans-parent, and sustainable regulatory framework, recognised internationally, which improves clinical safety and creates fair market access for manufacturers.
In contrast to Directives, Regulations are directly applicable and do not need to be trans-posed into national law. The MDR will therefore reduce the risks of discrepancies in interpretation across the EU market.
The MDR will replace the existing Medical Devices Directive 93/42/EEC (MDD) and Active Implantable Medical Devices Directive 90/385/EEC (AIMDD). The MDR was published in May 2017, marking the start of a four-year period of transition from the MDD and the AIMDD.
For Medical Devices (MDs) the transition period will end on 26 May 2021, the “Date of Application” (DoA) of the MDR.
During this period of transition, the Regulation will come into application gradually, starting with the provisions related to the designation of Notified Bodies and the ability of manufacturers to apply for new certificates under the Regulations.
To avoid market disruption and to allow a smooth transition from the Directives to the Regulations, several transitional provisions are in place. Some devices with certificates issued under the Directives may continue to be placed on the market [‘Placing on the market’ means the first making available of a device, other than an investigational device, on the EU market (Article 2 Section 28 of the MDR)] until 26 May 2024, and made available [‘Making available on the market’ means any supply of a device, other than an investigational device, for distribution, consumption or use on the Union market in the course of a commercial activity, whether in return for payment or free of charge (Article 2 Section 27 of the MDR)] until 26 May 2025.
What is changed?
In general, the MDR retain all the requirements of the Directives, while adding some new requirements of their own. Compared to the current Directives, the new Regulation emphasize a life-cycle approach to safety, backed up by clinical data.
The Regulation add more stringent rules for the designation of Notified Bodies. For national competent authorities and the Commission, they add more control and monitoring requirements. The Regulations clarify the obligations of manufacturers, Authorised Representatives, importers and distributors.
The MDR reclassifies certain devices and has a wider scope than the Directives. It introduces an additional pre-market consultation procedure for certain high-risk medical devices. For IVDs, the biggest change concerns the risk classification of in vitro diagnostic devices and the role of Notified Bodies. As a result, around 85% of all IVDs will need oversight from Notified Bodies, compared to 20% under the Directive.
The Regulations increase transparency, requiring the publication of information on devices and on clinical and performance studies related to their conformity. The new European Database for Medical Devices and In Vitro Diagnostic Medical Devices – EUDAMED – will play a central role in making data available and increasing both the quantity and quality of data (MDR Article 33 and IVDR Article 30).
EU - Timelines
Until May 2025, products certified under the Directives and products certified under the Regulations may coexist on the market. Both will have equal status under the legislation, and no discrimination in public tenders may take place.
A transition period is needed as the new Regulations require the designation of Notified Bodies. In addition, manufacturers need to meet more stringent criteria, particularly in terms of clinical and performance evaluation requirements.
The designation process for Notified Bodies, which may take 18 months or more, involves assessors from both national and European authorities. This means that the first Notified Bodies designated under the new Regulations might be established by the beginning of 2019. You can find the Notified Bodies designated under the MDR and IVDR, as well as the scope of devices for which they are designated, on New Approach Notified and Designated Organizations [‘NANDO’] (http://ec.europa.eu/growth/tools-databases/nando/). For more information refer to the contact points of the competent authorities (https://ec.europa.eu/health/md_sector/contact_en).
The rules for designating Notified Bodies are also more rigorous and add new requirements and responsibilities. The process of designating Notified Bodies will take up a significant part of the transition period, meaning that there will be limited time for manufacturers to have all their products certified before the respective DoAs.
This makes it unlikely that all devices available on the market will be certified under the new Regulations by the DoAs, especially if the designation of Notified Bodies takes longer than foreseen. To avoid market disruption and the unavailability of medical devices, manufacturers may, under certain conditions, continue to produce MDD/IVDD-compliant devices and place them on the market after the respective DoAs. These will be available for sale to end customers until 26 May 2025.