
Unique Device Identification (UDI)
The Regulations (EU) 2017/745 on Medical Devices and Regulation (EU) 2017/746 (“MDR”) on In Vitro Diagnostic Medical Devices (“IVDR”) introduce an EU identification system for medical devices based on a Unique Device Identifier (UDI).
The UDI system will facilitate easier traceability of medical devices, significantly enhance the effectiveness of the post-market safety-related activities for devices and allow for better monitoring by competent authorities.
The system applied to all medical devices except custom-made and performance study/investigational devices and is substantially based on internationally recognized principles, notably by using definitions that are compatible with those used by major trade partners.
Transition Timelines
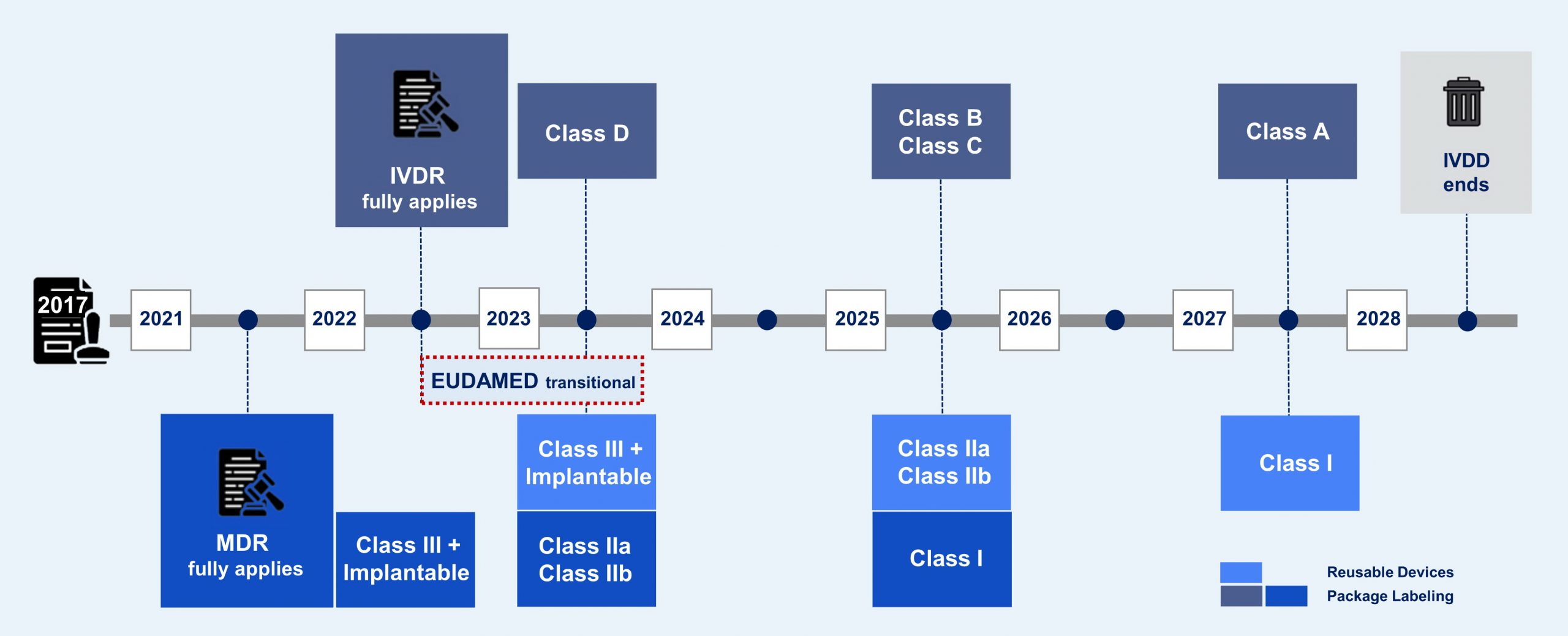
Article 27 MDR and Article 24 IVDR described that the UDI system shall consist of:
- the production of a UDI that comprises a UDI device identifier (‘UDI-DI’) specific to a manufacturer and a device, providing access to the information, and a UDI production identifier (‘UDl-PI‘) that identifies the unit of device production and if applicable the packaged devices, as specified in Part 0 of Annex VI;
- the placing of the UDI carrier on the label of the device or on its packaging or in case of reusable devices on the device itself (direct marking);
- the storage of the UDI by economic operators, health institutions and healthcare professionals, in accordance with the conditions laid down in paragraphs 8 and 9, respectively, of the Articles;
- the establishment of an electronic database for Unique Device Identification (the ‘UDI database’), which is part of the EUDAMED database, in accordance with Article 28 of MDR and Article 25 of IVDR.
The manufacturer shall thus assign a unique UDI to a device and to all higher levels of packaging before placing that device on the market except custom-made medical devices and performance study/investigational devices. The UDI carrier shall be placed on the label of the device and on all higher levels of packaging and in case of reusable devices on the device itself (direct marking). The manufacturer shall also ensure that the information, related to the device in question, referred to in Part B and Part A, Section 2, of Annex VI of the relevant Regulation, is correctly submitted to the European Database on medical devices (EUDAMED) as required by Article 27(3) of MDR and Article 24(3) of IVDR. The manufacturer shall also maintain unique UDIs for its devices.
The manufacturer shall assign to their devices, together with a UDI, also a Basic UDI-DI, which is not yet required by other jurisdictions. The Basic UDI-DI is the main key in EUDAMED and relevant documentation (e.g. certificates, declaration of conformity, technical documentation and summary of safety and clinical performance) and will also be the access key for device-related information entered in the database.

UDI meaning
The UDI is a series of numeric or alphanumeric characters that is created through a globally accepted device identification and coding standard. It allows the unambiguous identification of a specific medical device on the market. The UDI is comprised of the UDI-DI and UDI-PI. The unique identifier may include information on the lot or serial number and be able to be applied anywhere in the world.
The production of a UDI comprises the following:
Basic UDI-DI
The Basic UDI-DI is the main access key for device-related information in the EUDAMED database and it is referenced in relevant documentation, e.g. certificates (including certificate of free sale), EU declaration of conformity, technical documentation and summary of safety and (clinical) performance).
- It is intended to identify and connect devices with the same intended purpose, risk class and essential design and manufacturing characteristics.
- It is independent/separate from the packaging/labelling of the device and it does not appear on any trade item.
- Any Basic UDI-DI shall identify the devices (group) covered by that Basic UDI-DI in a unique manner.
UDI labeling
The manufacturer is responsible for complying with all UDI related requirements. This includes the assignment of the UDI (and Basic UDI-DI), the UDI (and Basic UDI-DI) registration the EUDAMED database and the placement of the UDI carrier on the label of the device or on its packaging or, in case of reusable devices, on the device itself (direct marking).
UDI registration
Systems and procedure packs shall undergo a UDI registration, as described in Article 29(2) of MDR.
Before placing on the market a system or procedure pack pursuant to Article 22(1) and (3), that is not a custom-made device, the system or procedure pack producer shall assign to the system or procedure pack, in compliance with the rules of the issuing entity, a Basic UDI-DI and shall provide it to the EUDAMED database together with the other relevant core data elements listed in the MDCG 2018-4 guidance document.
UDI issuing entities
The issuing entities operate a system for the assignment of UDIs. Following a call for applications launched at the end of 2018, the Commission has designated the following entities:
UDI appearance
The UDI Carrier, an automated Identification for Data Capture (AIDC) and human readable interpretation (HRI) representation of the UDI, shall be on the label or on the device itself and on all higher levels of device packaging.
In the event of significant space constraints on the unit of use packaging, the UDI carrier may be placed on the next higher packaging level. Higher levels of packaging shall have their own unique UDI. Please note that shipping containers shall be exempted from the requirement.
The UDI must appear in a plain-text version/human readable information (HRI) and in a form that uses AIDC technology. AIDC means any technology that conveys the unique device identifier or the device identifier of a device in a form that can be entered into an electronic patient record or another computer system via an automated process. The HRI consists of legible characters that can easily be read by people.
If there are significant constraints limiting the use of both AIDC and HRI on the label, only the AIDC format shall be required to appear on the label. For devices intended to be used outside healthcare facilities, such as devices for home care, the HRI shall, however, appear on the label even if this results in there being no space for the AIDC.
For single-use devices of classes I and Ila medical devices and class A and class B IVD medical devices packaged and labelled individually, the UDI carrier shall not be required to appear on the packaging but it shall appear on a higher level of packaging, e.g. a carton containing several (individually packaged) devices. However, when the healthcare provider is not expected to have access, in cases such as in-home healthcare settings, to the higher level of device packaging, the UDI shall be placed on the packaging (of the individual device).
For devices exclusively intended for retail point of sale, the UDI- Pls in AIDC shall not be required to appear on the point-of-sale packaging.
If the UDI carrier is readily readable or, in the case of AIDC, scannable, through the device’s packaging, the placing of the UDI carrier on the packaging shall not be required.
UDI-PI meaning
If a lot number, serial number, software identification or expiry date appears on the label, it shall be part of the UDI-PI. If there is also a manufacturing date on the label, it does not need to be included in the UDI-PI. If there is only a manufacturing date on the label, this shall be used as the UDI-PI.
The different types of UDI-Pls include serial number, lot number, software identification and manufacturing date and/or expiry date. The UDI-PI characteristics such as the lot or serial number shall be defined by the manufacturer. However:
- For active implantable devices, the UDI-PI shall include at least the serial number; for other implantable devices, the serial number or lot number.
- configurable device UDI-PI shall be assigned to each individual configurable device.
It is important to note that no UDI-PI information can be included in the UDI database.
UDI-DI changes
A new UDI-DI shall be required whenever there is a change that could lead to misidentification of the device and/or ambiguity in its traceability. In particular, a new UDI-DI shall be required in the case of any change of the following elements: name or trade name, device version or model, labelled as single use, packaged sterile, need for sterilization before use, quantity of devices provided in a package, critical warnings or contra-indications and CMR/Endocrine disruptors.
A UDI-DI shall be associated with one and only one Basic UDI-DI.
UDI obligations
According to the two Medical Devices Regulations, manufacturers shall be responsible for the UDI assignment and placement of the UDI carrier, the initial submission and updates of the identifying information and other device data elements in the EUDAMED database. Manufacturers shall update the relevant database record within 30 days of a change being made to an element, which does not require a new UDI-DI.
Distributors and importers shall verify that, where applicable, a UDI has been assigned by the manufacturer.
All economic operators and health institutions shall store and keep preferably by electronic means the UDI of the devices, which they have supplied or with which they have been supplied if those devices belong to class III implantable devices. Please note that the Commission may decide to adopt implementing acts to expand the scope of devices for which economic operators shall store and keep the UDI.
UDI on software
The UDI shall be assigned at the system level of the software.
Only software that is commercially available on its own and software that constitutes a device in itself shall be subject to that requirement.
The software identification shall be considered the manufacturing control mechanism and shall be displayed in the UDI-PI.
UDI requirements for software are laid down in Annex VI Part c of the two Medical Device Regulations.
https://ec.europa.eu/health/system/files/2021-04/md_mdcg_2018-1_guidance_udi-di_en_0.pdf