
NEWS - Letter 01 / 2022
New transition periods of the IVDR
As we can see from a recent press release, the EU has extended the transition periods of Regulation 2017/746 on in-vitro diagnostics (IVDR) in an urgent procedure. The European Council and the European Parliament adopted the European Commission’s proposal of October 14, 2021 without any changes. However, this does not change anything when the regulation comes into force. This remains May 26, 2022. However, the new deadlines give manufacturers and notified bodies a little more room to get IVD products through the conformity assessment procedure.
As a result, the new regulations could e.g. prevent IVDs from disappearing from the market en masse from May 2022.
In this article, you can find out which new deadlines apply, what they mean for you and what you should do now.
1. Overview of the new transition periods
a) What is new with the amending regulation of the IVDR?
In particular, the changes give manufacturers more time whose products require a Notified Body under the IVDR. Such products may still be manufactured and placed on the market after May 26, 2022. But beware! Some requirements of the IVDR still have to be met. You can find out more about this below.
b) Which products do the changes affect?
First of all, it is important that the new regulations only apply to existing products. These are devices that were (or will be) declared compliant before May 26, 2022 AND are classified as Class D, C, B, or A (sterile) under the IVDR. This means that all products will benefit from an extended transition period, except
• Class A products without sterile marking and
• new products for which a declaration of conformity will only be issued after May 26, 2022.
c) How long are the new deadlines?
How long the new deadlines are depends on the future risk class and whether a notified body was already involved under IVDD.
• There is no extended period for Class A devices, with the exception of sterile devices.
• These sterile Class A devices, along with Class B devices, will have the longest transition period for placing on the market: until May 26, 2027.
• For Class C products, the deadline is May 26, 2026 and
• For Class D products, the deadline is May 26, 2025.
• The so-called „sales regulation“ applies for a further year after the end of the aforementioned periods.
The figure below summarizes the regulations. Find out in the following section how this looks in detail for the individual cases.
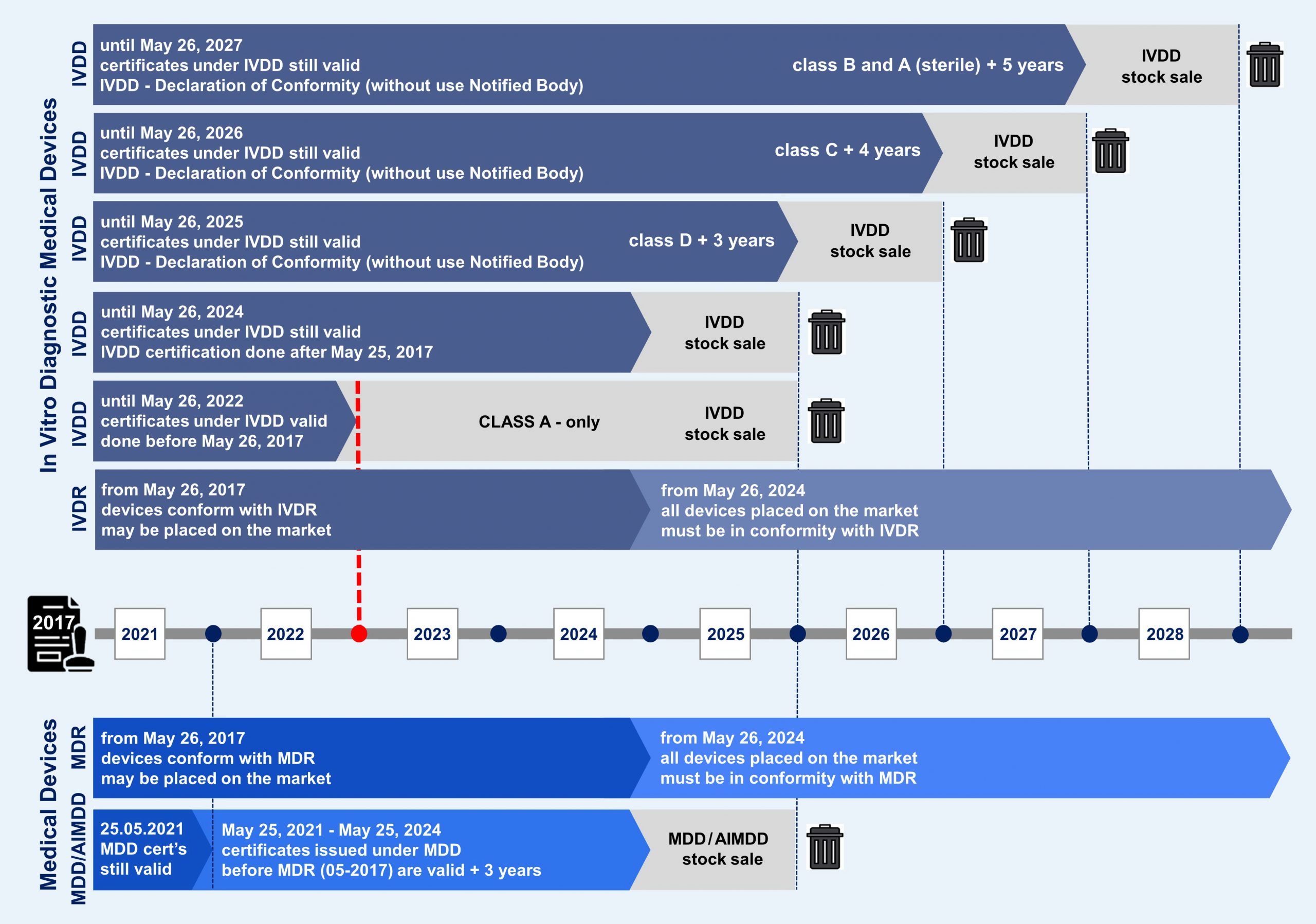
2. The new deadlines in detail
a) IVD for which a certificate was issued by a Notified Body under IVDD and also require a Notified Body under IVDR
For in vitro diagnostic devices for which a certificate has been issued by a Notified Body under IVDD, the deadline changes as follows in accordance with Article 110 paragraph (2):
• Certificates issued by Notified Bodies before May 25, 2017 according to IVDD remain valid until the date specified in the certificate. So there is no change for these products.
• Certificates according to Annex VI of the IVDD lose their validity on May 27, 2025 at the latest.
(before: 2024 – extension by 1 year)
• Certificates issued by Notified Bodies after May 25, 2017 in accordance with IVDD will lose their validity by May 27, 2025 at the latest. (before: 2024 – extension by 1 year)
What does that mean specifically?
Example: Let’s say you have declared conformity for a List B IVD with a Notified Body certificate according to Annex IV of the IVDD (Full Quality Assurance System). The certificate has an issue date before May 25, 2017. The validity date on the certificate should then be shown up to May 25, 2022 at the latest, because that is the requirement of the IVDD (Article 9 Para. 10). Anyone who does not receive a current certificate under IVDD from the notified body for their existing products by May 25, 2022 at the latest must declare conformity under IVDR. For this, manufacturers must involve a notified body accredited for the IVDR. With a certificate that was issued between May 25, 2017 and May 25, 2022, the validity of the certificate must end on May 26, 2025 at the latest (last possible date), otherwise it will become invalid according to IVDR regulation Art. 110.
The prerequisite for this case is that a notified body is or was already involved in the conformity assessment procedure under the IVD Directive 98/79/EC. Otherwise, no certificate from a notified body can exist.
Products that receive such a certificate are products that comply with Annex II, List A and List B or self-test, regardless of the conformity assessment procedure selected under the IVDD. So it doesn’t matter whether the product in question has received a certificate under IVDD via a complete quality assurance system, via the type examination or the design test in self-tests.
b) “Other IVDs” that have already been placed on the market under IVDD and require a Notified Body under IVDR
• The following new transitional periods apply to in-vitro diagnostics for which manufacturers under IVDD issued the declaration of conformity themselves before May 26, 2022 without involving a notified body and which under IVDR must undergo a conformity assessment procedure with a notified body in accordance with IVDR classification rules:
• May 26, 2025 for Class D IVD
• May 26, 2026 for Class C IVD
• May 26, 2027 for Class B IVD
• May 26, 2027 for Class A IVDs that are marketed sterile.
• Depending on the risk class of the IVD, these existing products may continue to be placed on the market after the IVDR’s validity date of May 26, 2022, provided that the products comply with the IVDD requirements and there are no significant changes to the design and purpose.
• However, manufacturers should note that even for devices for which the proposed transition periods would apply, the requirements of the IVDR for post-market surveillance (PMS), market surveillance, vigilance and registration of economic operators and devices must be complied with.
What does that mean specifically?
Assuming that the product has been or will be issued with a declaration of conformity according to Annex III under the IVDD before May 26, 2022 without a notified body. The question then arises under which class the product will fall under the IVDR in the future.
I) If the product falls into Class D in the future, you may still manufacture and market these products until May 26, 2025, but you may no longer make any significant changes. After that, you or the distributor may provide and commission the product for another year until May 26, 2026, which is colloquially referred to as the “sales rule”.
II) If the product falls under Class C in the future, manufacture and placing on the market is permitted until May 26, 2026. Significant changes are not permitted.
III) If the product falls under class B or A (sterile) in the future, manufacture and placing on the market are permitted until May 26, 2027, which is the longest transitional period. Significant changes to the product are also not permitted here.
c) No extension for “normal” (non-sterile) Class A devices and “new” IVDs
However, the extensions proposed by the Commission should NOT apply to two product groups:
• Normal” (non-sterile) Class A products
• „New“ IVD
In any event, as of May 26, 2022, if the device falls under Class A and is not a sterile device, manufacturers must demonstrate compliance with the IVDR and meet all applicable IVDR requirements. You cannot use an extended transition period. With no Notified Body required, it is entirely up to the manufacturer’s resources and timing to achieve this goal.
The extended deadlines for „new“ in-vitro diagnostics also do not apply. As such, the Commission considers IVDs whose conformity has not been declared under the IVDD.
Manufacturers must also provide proof of conformity for these products in accordance with IVDR from May 26, 2022.
d) Laboratory Developed Tests (LDT)
There is now also a little more leeway for products that are produced and used by medical institutions themselves. This is intended to relieve the burden on health facilities such as laboratories, which have been badly hit by the corona pandemic.
But be careful, the following requirements remain unaffected:
(3) Demonstrating compliance with the essential safety and performance requirements shall also include a performance evaluation in accordance with Article 56.
(4) Products manufactured and used in healthcare facilities are deemed to have been put into operation.
(5) With the exception of the relevant essential safety and performance requirements set out in Annex I, the requirements of this Regulation shall not apply to devices manufactured and used solely within healthcare facilities located in the Union, provided that all of the following conditions are met
Article 5 (5) sentence 1, sentence 2 letter a) and sentences 3 to 5 also remain unchanged.
From May 26, 2022, all Lab Developed Tests (LDT) in the EU must meet the basic safety and performance requirements in accordance with Annex I IVDR, including the performance evaluation, and may not be handed over to another legally independent institution – legal requirements that apply in Germany are already anchored in the Medical Devices Act (MPG).
Article 5 (5) of the IVDR provides for longer periods of time for LDT, especially with regard to the sub-items of sentence 5. For the implementation of sub-points b) and c) as well as e) to i), the deadline is now May 26, 2024. For letter d), the deadline is May 26, 2028.
Letter d) has the longest period. This says:
„In its documentation, the healthcare facility provides a justification for the fact that the specific needs of the patient target group cannot be satisfied or cannot be satisfied at the indicated level of performance by a similar product on the market;“
This requirement should ensure that there is no competition between LDTs and commercial products on the market, as they are regulated differently. The EU has now placed this point at the very end of the transition period on the grounds that laboratories can only compare the products on the market with their own product if all IVD products on the market are also approved under IVDR and registered in EUDAMED.
For which the EU grants a delay in LDTs
The following items of Article 5(5) will be deferred for two years until May 26, 2024:
• Quality management system
Letter b) refers to the requirement for a suitable quality management system, which would at least have to take into account the development and manufacture of IVDs from ISO 13485 in order to meet the state of the art.
• Quality assurance in the laboratory
Letter c) requires compliance with the requirements of ISO 15189 for quality assurance in the laboratory and compliance with national accreditation regulations such as the RiliBÄK in Germany (guideline of the German Medical Association for quality assurance in laboratory medicine).
• Provision of information
Letter e) Provision of information to authorities on manufacture, modification and use
• Public statement
Letter f) Declaration of compliance with Annex I of the IVDR
• Documentation
Letter g) Documentation on production, design and performance, i.e. the “product file” so to speak
• Proof of production according to specifications (batch record)
Letter h) Proof of manufacture according to the specifications in the „Product File“
• Serious Incidents and Corrective Actions
Letter i) Collecting and evaluating experience in clinical use and deriving corrective measures
e) EUDAMED
According to the current status, the deadline for an EUDAMED registration of IVDs is November 26, 2023.
This results from Article 26 paragraph 3 IVDR and Article 113 IVDR, which refers to it. Art. 113 IVDR also points out that IVDs must be registered in EUDAMED 18 months after the IVDR has come into force. If the database is fully functional on May 20, 2022, the deadline for registration is November 26, 2023.
3. What does not change
The IVDR applies from May 26, 2022. This means that the obligations to monitor products on the market (Articles 78 to 81) as well as vigilance (Articles 82 to 87) and monitoring by the competent authorities also apply, regardless of the transition periods. Thus, the entire Chapter VII of the IVDR is to be applied, even if the products are still placed on the market under IVDD. In addition, economic operators must register in EUDAMED.
However , the requirements of this Regulation relating to post-market surveillance, market surveillance, vigilance, registration of economic operators and of devices shall apply to devices referred to […] instead of the corresponding requirements in Directive 98/79/EC.
Proposal of the EU Commission of October 14, 2021 to adapt the IVDR (EU) 2017/746
The Notified Body that issued certificates under the IVD Directive will continue to be responsible for monitoring conformity during the transitional period.
It is also important that no significant changes are made to the products or their intended purpose during the transition period, because then manufacturers must apply the IVDR immediately and in full.
4. Background to Changes
The IVDR came into force on May 25, 2017 and will be binding from May 26, 2022. The new deadlines do not change that. As a result, a large part of the declarations of conformity issued under the IVDD would have become invalid from May 2022.
However, on October 14, 2021, the EU Commission submitted a proposal for an amendment to the IVDR to the Council and Parliament in order to give manufacturers more time to go through the conformity assessment procedure. The resulting amending ordinance went through the legislative process in a very short time.
Reasons
The Commission justified its proposal primarily with the effects of the corona pandemic: This ties up a lot of resources, making it almost impossible to implement the necessary measures quickly.
But the current effects of the MDR on the Medtech-Market should also play a role. A product clear-cut such as that caused by the MDR should be avoided for in-vitro diagnostics. Various actors have been sounding the alarm for months.
• Numerous stakeholders from politics, the Medtech-Industry, notified bodies, medical professionals, scientists and non-profit organizations have warned of the dangers of IVDR deadlines being too short.
• On May 11, 2021 and June 15, 2021, various parties and the Council of Health Ministers contacted the Commission to obtain a new regulation.
In its proposal, the commission recognized how dramatic the situation is:
• There are currently only six Notified Bodies across Europe that can deal with the IVDR (under the IVDD there were 18).
• A notified body must now be involved for significantly more in-vitro diagnostics: Up to now, the quota was around 8% of the in-vitro diagnostics on the market. Under the IVDR, a notified body would have to be involved in almost 78% (a total of around 24,000 IVDs).
• According to a survey by MedTech Europe, if the deadlines were not extended, around 22% of in vitro diagnostics would disappear from the market from 2022 (currently around 40,000 IVDs are on the market, i.e. 9,000 would disappear).
• However, Covid-19 in particular has shown how important reliable in-vitro diagnostics are.
• Approximately 70% of clinical decisions are made using in vitro diagnostics.
5. What the changes mean for manufacturers
• It is important for manufacturers to continue to adhere to their implementation plan so that their IVDs become IVDR-compliant in good time.
• Due to the scarce resources, manufacturers should not let time pass unnecessarily.
• Manufacturers should be clear about the risk class of their IVD. Not only does the conformity assessment procedure depend on this, but also the transition period.
6. Conclusion
The IVDR will apply from May 26, 2022. The new regulations in the IVDR will not change that. However, the majority of IVD manufacturers now have more time to provide the necessary proof of conformity for their products. In view of the few notified bodies that have existed for the IVDR so far and the generally tense situation due to the corona pandemic, this should bring great relief for many – and prevent important IVD products from disappearing from the market. For medical laboratories, too, some requirements only apply with a time delay. Nevertheless, they must also meet the basic safety and performance requirements for in-house products or lab-developed tests.
Source:
https://ec.europa.eu/commission/presscorner/detail/en/ip_21_6965
Johner Institute – Neue Übergansfristen IVDR